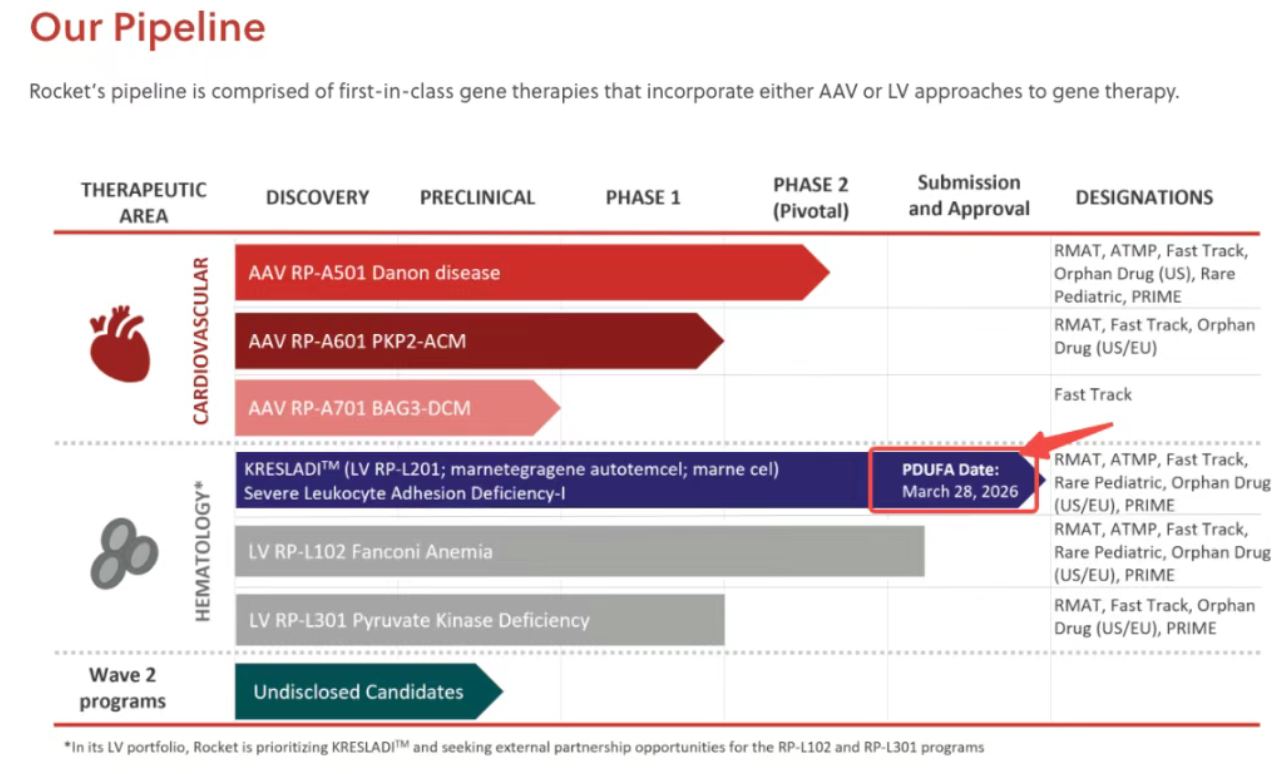
3月27日,距离美国 FDA 对 Rocket Pharma 的核心基因疗法KRESLADI™(marnetegragene autotemcel)给出最终裁决的PDUFA日期(2026年3月28日),仅剩最后的48小时冲刺窗口。
关于KRESLADI
LAD-I (Leukocyte Adhesion Deficiency Type I,I型白细胞黏附缺陷症)是一种极为罕见、极具破坏性且致命的遗传性免疫功能缺陷疾病。由于 ITGB2 基因发生突变,患者体内的白细胞无法表达CD18蛋白,导致白细胞无法正常黏附并穿透血管壁到达组织感染部位。这意味着,患儿对哪怕是最微小的细菌或真菌感染都毫无招架之力。其典型临床表现包括出生后脐带延迟脱落、反复发生危及生命的皮肤和黏膜感染。
在没有进行异基因造血干细胞移植(这是以往唯一的治愈希望,但面临供体难寻和严重的移植排斥风险)的情况下,重度LAD-I患儿在儿童期死亡的概率几乎是100%。
面对这一残酷的“孤儿病”,KRESLADI提供了一种具有颠覆性的一次性治愈方案。
-
作用机制:KRESLADI是一种基于慢病毒载体(LV)的体外(ex vivo)自体基因疗法。它首先提取患者自身的造血干细胞,在高度受控的实验室环境中,利用慢病毒载体将功能正常的 ITGB2 基因导入这些细胞中,然后再将这些经过“基因修正”的细胞回输给患者。这些细胞随后会在患者体内重构免疫系统,恢复白细胞的抗感染能力。
-
临床数据:在1/2期全球临床试验中,KRESLADI展现出了很好的疗效。数据显示,接受治疗的患者在12个月时的总体生存率达到了100%,所有主要和次要终点均顺利达成。该疗法不仅显著减少了严重感染的发生频率,还极大改善了患者的皮肤病变情况并恢复了正常的伤口愈合能力。
波折的监管之路
回顾KRESLADI的获批之路,可谓是一波三折。这也是近年来众多前沿 CGT 在面对FDA严格监管时的一个缩影。
-
审查延期:KRESLADI的 BLA 在2023年10月就获得了FDA的优先审评资格。然而,FDA为了有充足时间审查Rocket提交的补充信息,将原定于2024年初的PDUFA日期推迟至了2024年6月底。
-
遭遇CRL:2024年6月28日,FDA向Rocket下发了一封完整回应函(CRL),要求提供更多关于化学、制造和控制(CMC)的信息。对于基因疗法而言,由于其具有“产品即工艺”的特殊性,如何保证复杂生物制剂在生产过程中的一致性、纯度以及放行检测的标准化,往往是跨越监管的最难关卡。这一挫折曾一度引发资本市场的担忧。
-
强势回归:Rocket并未气馁。公司在随后进行了深度的战略重组与管线优先级调整,集中核心资源全力解决FDA指出的CMC缺陷。在与监管机构进行密切沟通后,FDA于2025年10月正式接受了KRESLADI的BLA重新提交,并将其最新的PDUFA日期锁定在了2026年3月28日。这一关键进展极大程度上消除了产品在安全性或疗效数据上的风险障碍,将其主要考量重新聚焦于最终的生产合规性上。
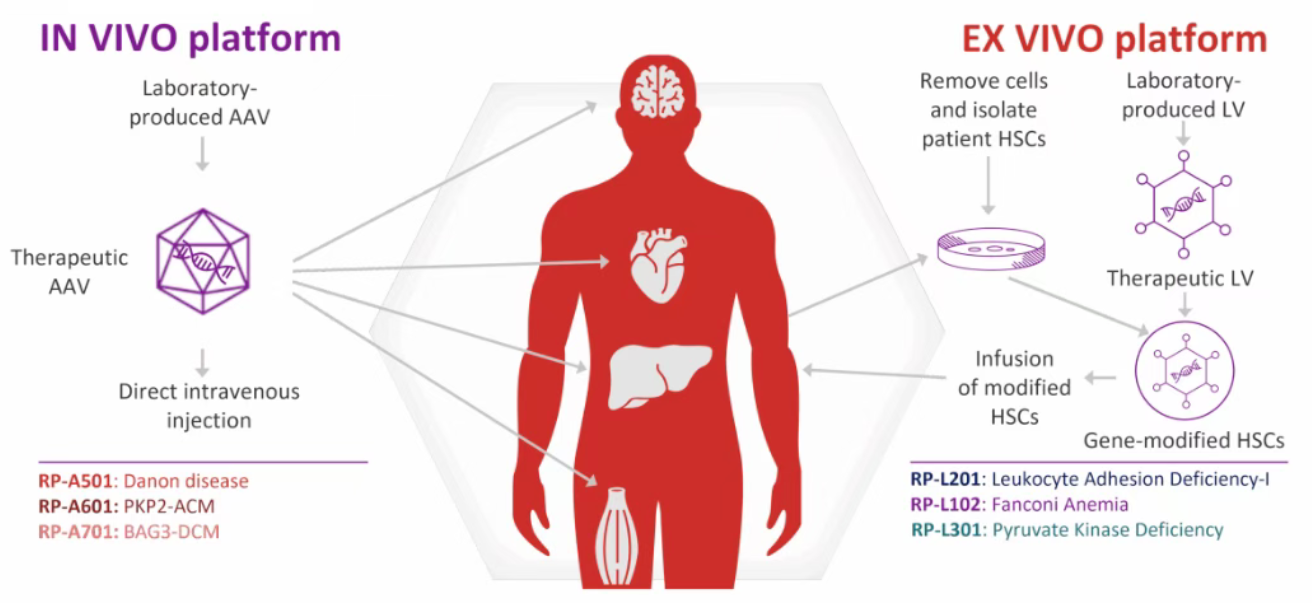
Rocket Pharma的研发平台
Rocket Pharmaceuticals 的多平台协同策略使其能够针对疾病的根本原因选择最匹配的递送技术:对于实体器官(如心脏),采用体内AAV平台直接递送基因;对于血液和免疫系统疾病,则采用体外LV平台对造血干细胞进行彻底改造。这一双引擎模式构成了其产品管线能够覆盖多种复杂、高度未满足临床需求罕见病的核心竞争力。